Elektrochemické HPLC detektory
Elektrochemické detektory se používají k detekci látek, které jsou schopné elektrochemické reakce, probíhající na fázovém rozhraní elektroda - roztok (mobilní fáze). Množství aplikací pro elektrochemickou detekci není příliš velké, ale látky, na které se aplikuje elektrochemická detekce, reprezentují velké množství důležitých léčiv, polutantů a přírodních produktů.
Elektrochemické detektory měří určitou elektrickou veličinu (elektrodový potenciál, proud, kapacita) vyvolanou průchodem látky průtokovou celou detektoru, ve které jsou umístěny elektrody s vloženým pracovním napětím nezbytným k průběhu elektrochemické reakce a to v systému dvouelektrodového nebo tříelektrodového zapojení (obrázek č. 1). Jako další proměnná veličina se zaznamenává čas. Měřený elektrický signál je úměrný látkovému množství detekované složky a u elektrochemických detektorů se tedy sleduje závislost mezi elektrickou veličinou a koncentrací sledované složky. Nejčastěji je využívaná elektrochemická reakce redoxního systému Ox + ne- -> Red a podle podmínek měření můžeme rozdělit elektrochemické detektory na několik typů. Je-li elektrochemický článek v termodynamicky rovnovážném stavu (faradický proud je nulový, I = 0) mluvíme o potenciometrii, je-li v kinetickém stavu (faradický proud není nulový, I ¹ 0) - elektrolytické metody, kdy článkem protéká proud a jedním směrem probíhá elektrolýza. U elektrolytických metod se uplatňují čtyři závisle proměnné veličiny: potenciál elektrody (E), proud (I), čas (t) a koncentrace elektroaktivní látky (c). Podle podmínek měření elektrolytických metod se rozdělují elektrolytické metody na metody potenciostatické, kdy se měří při konstantním potenciálu pracovní elektrody a metody amperostatické, kdy se měří za konstantního proudu.
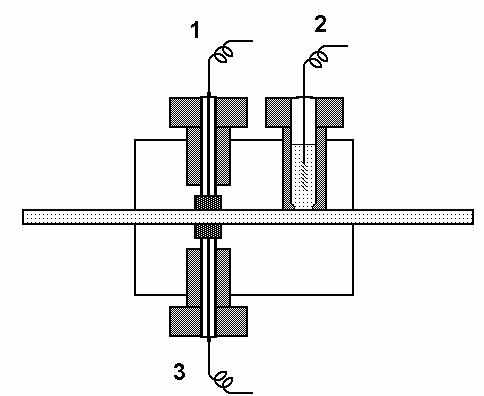
Obr. č. 1 Schéma elektrochemického detektoru
|
1. pracovní elektroda
2. pomocná elektroda
3. referenční elektroda
Amperometrické (polarografické) detektory měří proud vyvolaný průchodem redukované nebo oxidované látky průtokovou celou detektoru. Jako měrné elektrody se u amperometrických detektorů používají tuhé měrné elektrody zhotovené ze skelného uhlíku, grafitových vláken, zlata, platiny, mědi či jiného kovu. Povrch všech těchto materiálů se však zanáší produkty oxidace či redukce a nečistotami z mobilní fáze nebo vzorku a toto pak vyžaduje čištění těchto elektrod. U starších typů detektorů se toto čištění provádí mechanickým očištěním měrné elektrody po demontáži cely detektoru. U novějších typů se čištění provádí několikerým způsobem: otočení polarity elektrodového systému, čímž dojde k rozpouštění vyloučených látek na povrchu elektrody, nebo vkládáním střídavého napětí na elektrodu (pulsní technika).
Elektrické zapojení je u většinou stejné, liší se pouze svým geometrickým uspořádáním. Mobilní fáze omývá povrch pracovní elektrody a v tomto případě elektrochemická reakce probíhá pouze ve velmi tenké vrstvě (oxidačně-redukční reakci na povrchu elektrody podléhá méně než 10 % přítomného analytu) a konstrukce měrných cel má několik technických řešení. U válcové měrné cely eluát prochází kapilárou ve středu matky, ve které je umístěn držák s měrnou elektrodou, při konstrukci "wall-jet" eluát vytéká z trysky a omývá přímo měrnou elektrodu. Průtokové měrné cely lze snadno miniaturizovat pro práci s mikrokolonami nebo kapilárními kolonami. Vnitřní objem průtokových cel se může pohybovat kolem 1 nl. Kromě těchto dvou typů měrných cel byla popsána kombinace obou kontrukcí.[1]
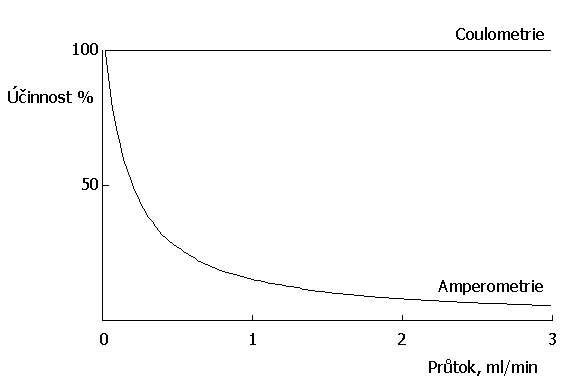
Jako srovnávací elektroda se používá kalomelová nebo argentchloridová elektroda. V těchto případech je účinnost elektrochemické reakce limitována dvěma faktory. Prvním faktorem je difuse elektroaktivní komponenty k povrchu pracovní elektrody, kde probíhá vlastní reakce a druhým faktorem je rychlost průtoku mobilní fáze. V důsledku toho je signál obdržený z amperometrického detektoru závislý na průtoku mobilní fáze, kdežto signál obdržený z coulometrického detektoru nikoliv (obrázek č. 2). Účinnost amperometrické elektrody je silně závislá na ploše povrchu znečištění díky elektrodepozicím a adsorpci, která na povrchu elektrody probíhá, z tohoto důvodu tudíž klesá i její analytický signál a detektor neposkytuje nikdy stejný analytický signál pro stejné analytické koncentrace analytu.
Obr. č. 2 Účinnost elektrochemické reakce v závislosti na průtoku mobilní fáze
|
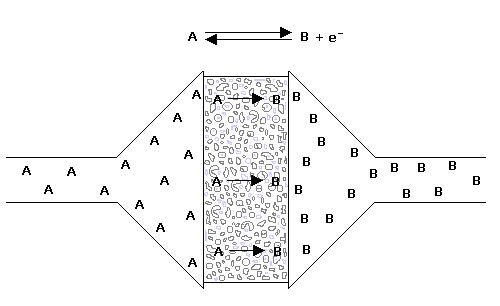
Coulometrické detektory měří náboj potřebný k oxidaci či redukci celkového množství látky při jejím průtoku měrnou celou detektoru a dosahuje se tak vyšší citlivosti detekce než u amperometrických detektorů. Účinnost elektrochemické reakce je možné zvýšit použitím tzv. elektrody fritového typu, kdy mobilní fáze protéká porézní grafitovou pracovní elektrodou. Výhodou této coulometrické elektrody je její vysoká účinnost, stabilita (snižuje se poměr signálu k šumu) a selektivita. Coulometrická elektroda tohoto typu má daleko větší povrch proti klasické elektrodě a oxidačně-redukční reakci na povrchu elektrody podléhá více jak 90 % přítomného analytu.
Obr. č. 3 Porézní grafitová pracovní elektroda fritového typu (firma ESA, Inc., USA)
|
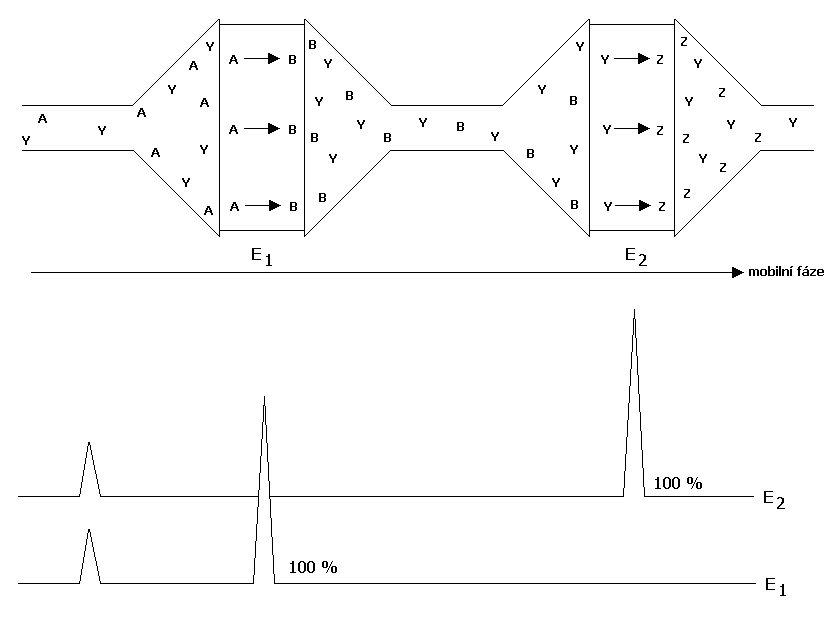
Další výhodou coulometrické elektrody je možnost zvýšení selektivity zapojením dvou a více elektrod v sérii na nichž je vloženo různé napětí. Protože se analyt na coulometrické elektrodě podrobuje 100 % elektro-lýze, efluent neobsahuje již žádnou elektroaktivní komponentu, která by mohla podléhat elektrochemické reakci při daném vloženém napětí. V případě, že existují v daném systému dvě separované komponenty lišící se svými elektrochemickými vlastnostmi (půlvlnným potenciálem alespoň o 60 mV), je možné je rozlišit voltametricky. Zapojením čtyř až šestnácti coulometrických elektrod je možné získat detektor obdobný detektoru diodového pole tzv. CoulArray (ESA, Inc., USA). K odstranění nečistot z mobilní fáze je možné použít tzv. guard celu (porézní grafitová elektroda, na kterou je vloženo určité napětí), která odstraňuje elektroaktivní nečistoty před vstupem do injektoru, analytické kolony a analytické cely.
Obr. č. 4 Rozlišení dvou elektroaktivních komponent lišící se svým půlvlnným potenciálem
|
Elektrochemické detektory dosahují vysoké citlivosti a jsou srovnatelné s citlivostí fluorimetrických detektorů. Elektrochemická detekce je kompatibilní jak s reverzními tak normálními fázemi. Protože však musí být mobilní fáze vodivá, má reverzní fáze v tomto před normálními fázemi výhodu a používá se proto ve větším měřítku. Vyšší požadavky se však kladou na čistotu mobilní fáze (voda a aditiva /pufry/ mobilní fáze, nízký obsah kovů) a dokonalost odvzdušnění (zamezit přítomnosti zejména rozpuštěného kyslíku), což je podmínka k dosažení stabilní základní linie a reprodukovatelnosti výsledků. Z aditiv mobilní fáze jsou vhodné fosfáty, octany a citráty, použití aminů by mělo být v co nejmenší míře. Vodivost mobilní fáze se může zvýšit přídavkem chloristanů (0,05 M), obsah organické složky (zejména methanolu) ve vodně-organických mobilních fázích by měl být co nejmenší. Přítomnost kyslíku a kovů (může docházet k "vymývání" kovů z chromatografického systému) v mobilní fázi může mít za příčinu významné pozadí a tudíž i zvýšený šum a drift základní linie. Snižení obsahu kyslíku bylo dosaženo přídavkem siřičitanů do mobilní fáze.[2] pH mobilní fáze má významný vliv na průběh voltamperometrické křivky a může být tak zcela nekompatibilní s pH mobilní fáze.
Výběr potenciálu detekce je obvykle kompromisem mezi citlivostí, stabilitou signálu a selektivitou založenou na faktu, že se zvýšením potenciálu detekce dojde ke zvýšení odezvy, ale současně dojde ke zvýšení šumu a driftu základní linie a snížení selektivity.
[1] Elecrochemical Detection in HPLC, Spark Holland B.V., Emmen, The Netherlands 1987.
[2] van Oort W.J., den Hartigh J., Driebergen R.J. in Electrochemical detectors (Ryan T.h. ed.), Plenum Press, New York 1984.
Last modified: